Integrating different genome assemblies
Kirill V. Romanenkov
DESCRIPTION
Genome assemblies of short reads (100-150 bp) are frequently fragmented and contain errors. The usual practice at the solution of the genome assembly problem in the case of de novo assembly consists in running several assemblers with different parameters, and then choosing the best option in accordance with some metrics. However recent studies show that situation in which some assemblers performed better on one of the metrics compared with other programs, and also concede to them in another metric is fairly common. [1].
Thus, different genome assembly tools could be used together to improve contiguity and accuracy and the problem of using strong points of each assembly remains rather challenging. A number of software tools aim to implement the strategy of symmetrical merging different genome assemblies, like MIX [2] and CISA [3]. These programs are implemented in interpreted languages and therefore combing even bacterial genome assemblies becomes a resource intensive task. Moreover, in some cases the quality of merged assembly raises certain questions.
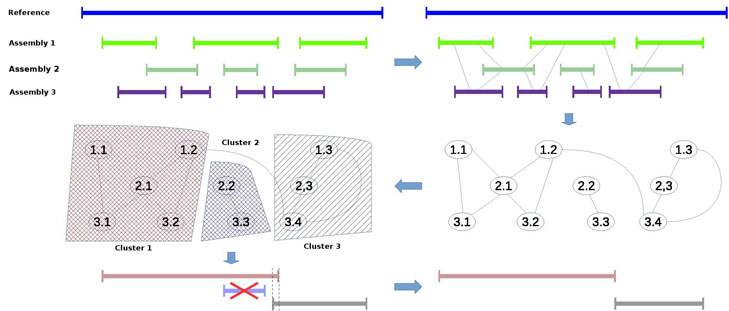
Due to the weaknesses of the above programs a new assembly integration method was proposed (GAR - Genome Assembly Refinement [4]). The algorithm consists of the four major phases:
- aligning each contig against the entire set of contigs;
- building and clustering an unordered weighted contig graph;
- merging contigs in each cluster using a greedy algorithm;
- removing end-to-end overlapping in obtained contigs and excluding from the final set contigs contained into other contigs.
The figure shows the steps of integrating three different genome assemblies with proposed method.
Our approach GAR automaticaly produces the integrated assembly of results of different genome assembly tools. To obtain a higher quality assembly a user should submit at least three sets of contigs in FASTA format, which are the outputs of different assemblers. If sets number is less than three, the program will not work.
User should run the assemblers on his own to obtain datasets to be uploaded to GAR. The source code of GAR is available at https://bitbucket.org/kromanenkov/gar.
|