On the basis of spectral-statistical approach [3, 4]
to identify reliable and significant heterogeneities in DNA sequences a technology was designed for search of latent tandem repeats in genomes. The theoretical maximum level of divergence of the of tandem repeat pattern copies being found is 50%.
A set of programs was developed which is universal because it works equally well with all kinds of periodicities: micro- , mini-, macro- and mega-satellites. The former are of length of pattern from 2 to 100 and the latter are of length of pattern of several thousand nucleotides (bps) [1, 2]. The computer technology is based on a model of evolution of a tandem repeat by means of successive duplications of adjacent copies of the text pattern.
Provided the true length of the period is a priori unknown, the revealing of hidden periodicities is a difficult task from the algorithmic point of view. In order to find fuzzy periodicities (the maximum level of divergence of 50%) the methodology [3] of searching of highly significant (at a level of α = 10-6) heterogeneities was selected. To confirm the significance of latent tandem repeats with small number of copies (in case of insufficient statistical material) a special quantitative criterion was used which reflects the quality [3, 4] of the character preservation of the pattern of periodicity.
For checking sequence heterogeneity in the found fragments, in general, two characteristics are used − the value of pattern preservation level pl (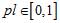 ) and the parameter HL indicating significant deviation from homogeneity (at the level of α = 10-6) on the test period. The maximum value of pl indicates the true length of period, provided that the parameter HL ensures heterogeneity of the nucleotide sequence on such a period lenght when the number of copies is more than 20. When the copy number is not exceeding 20, the only one characteristics - preservation level value pl of periodicity pattern is used. It should not be less than 0.625.
) and the parameter HL indicating significant deviation from homogeneity (at the level of α = 10-6) on the test period. The maximum value of pl indicates the true length of period, provided that the parameter HL ensures heterogeneity of the nucleotide sequence on such a period lenght when the number of copies is more than 20. When the copy number is not exceeding 20, the only one characteristics - preservation level value pl of periodicity pattern is used. It should not be less than 0.625.
The method of calculating of the pattern preservation level pl(L) consists of following steps. The analyzed sequence of length n consisting of the letters of the alphabet A = {a1,…,aK} is divided into substrings of length L (the final substring may have a smaller length). If n is the length of the analyzed sequence, then the number of substrings 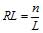 is called the test exponent for the test-period L. This division by substrings allows us to calculate the frequency of occurrence of the i-th letter of a nucleotide sequence alphabet in the j-th positions of a test-period. By the matrix of frequencies
is called the test exponent for the test-period L. This division by substrings allows us to calculate the frequency of occurrence of the i-th letter of a nucleotide sequence alphabet in the j-th positions of a test-period. By the matrix of frequencies 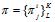 one can calculate the value of pattern preservation level:
one can calculate the value of pattern preservation level:
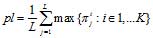
and the value of spectral-statistic parameter HL:
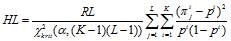 ,
,
Here pi − is the frequency occurrence of the i-th letter of the alphabet A in the analyzed sequence. In fact, HL is a normalized Pearson Χ2-statistics.
Chromosomal DNA is scanned repeatedly by using sliding window technique, the window length is twice the length of the test period, and the step of displacement of the window is variable, depending on the length of the test-period. The number of passes of the scan depends on the length of range of test-periods specified by the user (the default range is from 2 to 10 bps). These values are chosen in order to provide the reasonable calculation time for the sequence specified by the user. When expanding the range of lengths of test-periods the search procedure significantly slows.
If the value of preservation level for two equal length adjacent areas is not less than 0.875 (i.e. these areas differ by no more than a quarter of a point), we believe these sites are blurred repeats with multiplicity 2. Then, the overlapping portions of the same length of the test period have to be analyzed to form the merger, which represents a tandem repeat of higher multiplicity covered by a set of pairwise similar sites in the given chromosomal region. Special efforts are being made to clean up the left and right borders of area to find the true start and end positions related to the periodicity. Technology of the merges and removals of fragments is repeated many times with different parameters and conditions.
